Zygotic genome activation
|
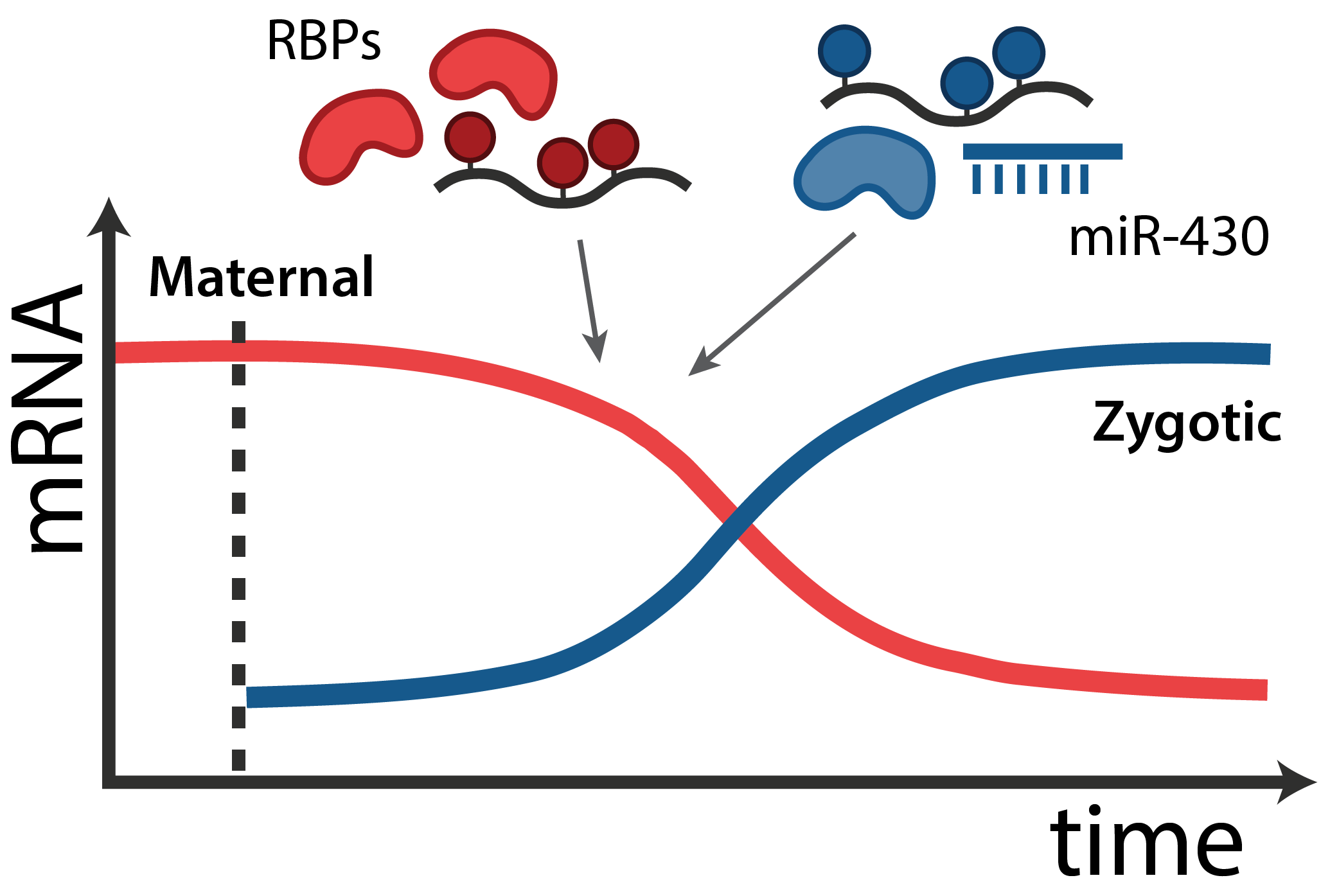
In all animals, maternal mRNAs and proteins deposited in the egg direct the initial stages of development and the embryos genome is transcriptionally silent. This set of “instructions” - called the maternal contribution - is fundamental to the development of every organism.
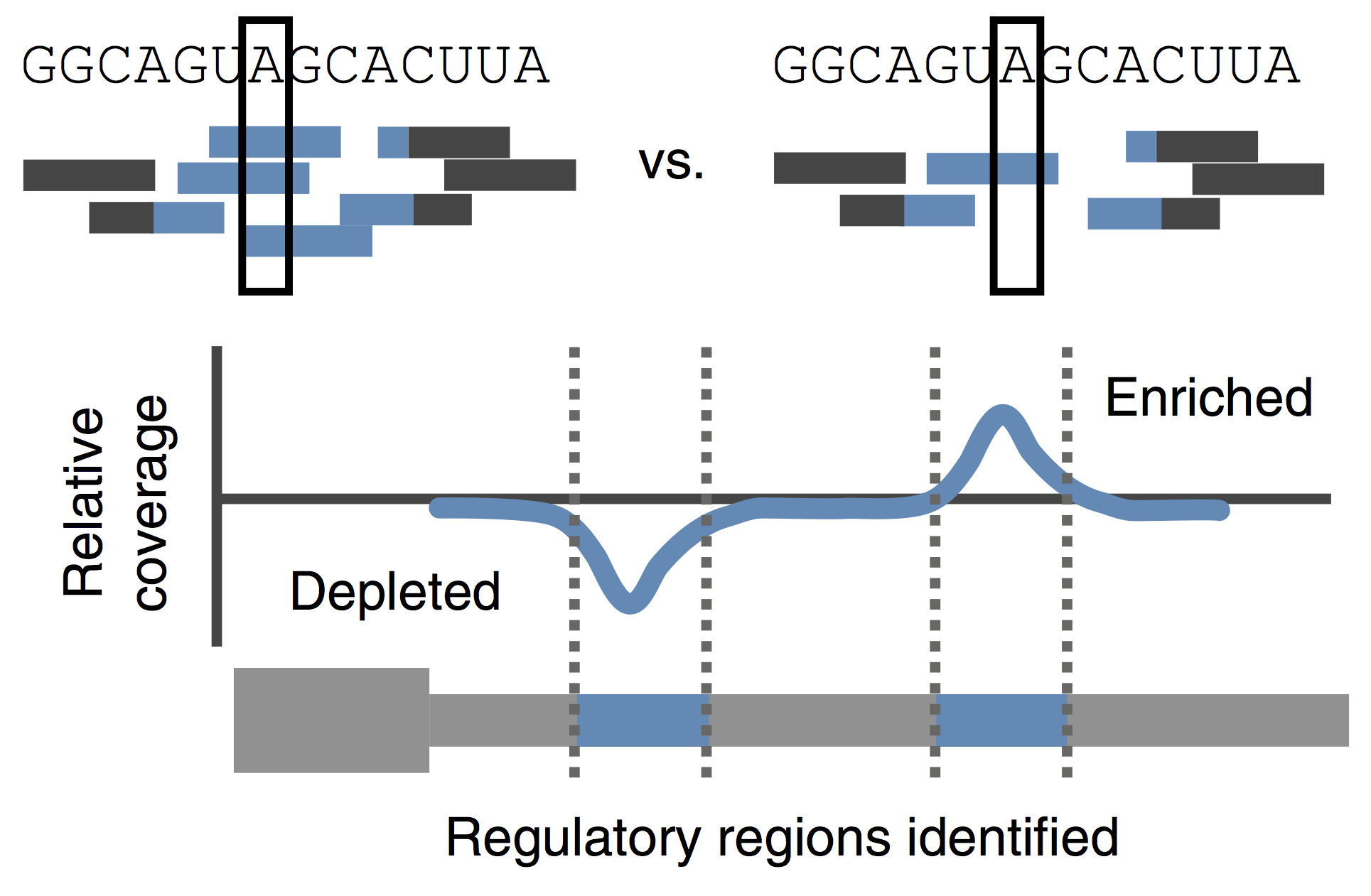
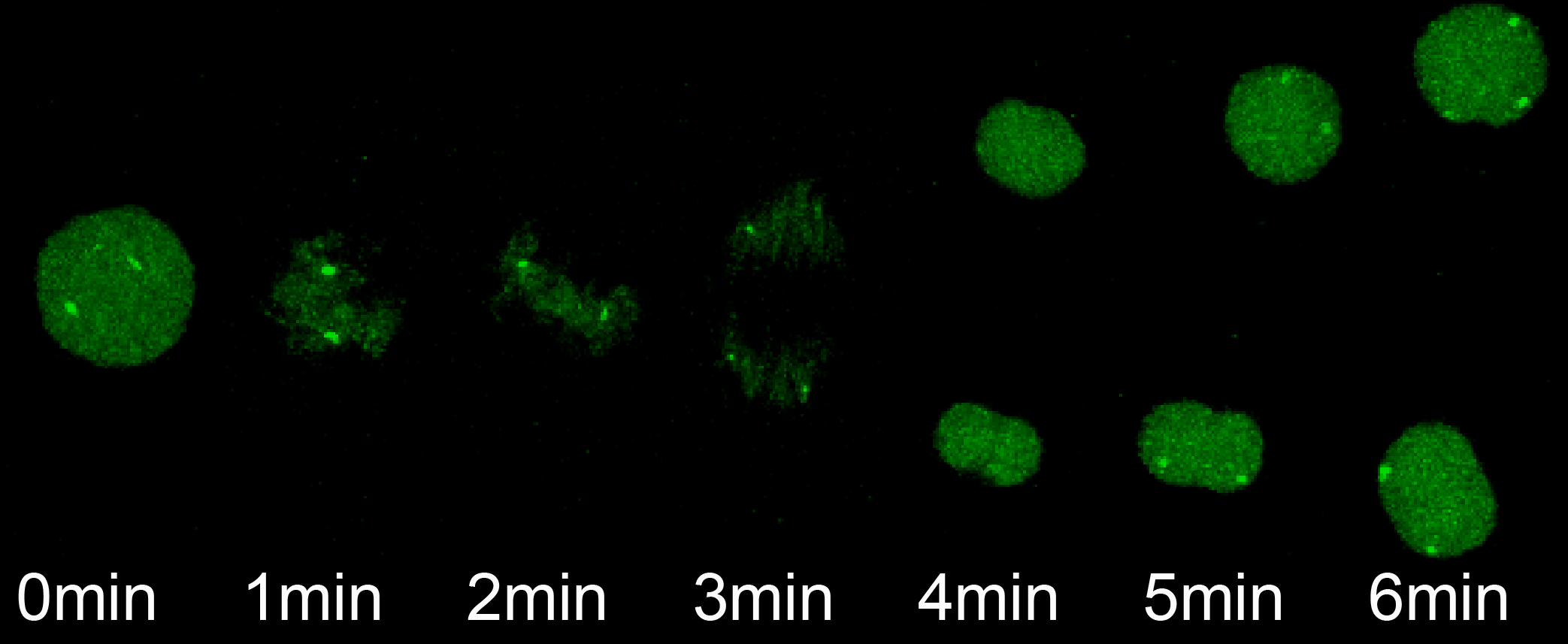
The transition from maternal-to-zygotic nuclear control represents a key event in developmental reprogramming. This process requires chromatin remodeling to establish transcriptional competency, maternal transcription factors to specifically activate the new transcriptional program and the clearance and degradation of maternal products. We are interested in understanding how these molecular events license the genome for activation using the genetic tools available to the zebrafish system in combination with biochemical and high throughput genomic approaches.
We have recently identified three key maternal transcription factors – Nanog, SoxB1 (Sox2) and Pou5f3 (Oct4) – as being widespread regulators of gene activation during this transition in zebrafish (Lee, Bonneau et al Nature 2013). Loss of these factors results in complete developmental arrest and failure to activate ~ 80% of zygotic genes. We are now interested in understanding the molecular mechanism by which these factors direct activation and mediate genome competence during this fundamental transition in biology.